

A Q&A guide to stability storage
This back to basics Q&A article on stability storage and testing has been put together by our expert team.
If you are reading this article, then you may also be interested in our new webinar series.
Following feedback from clients, we have recently curated a series of webinars designed for those working in the pharmaceutical, medical device and life sciences sectors with responsibility for designing and managing stability studies. If would like to gain some key insights into stability study design see our upcoming webinar series.
To find out more about how Q1 Scientific can help you with your stability storage and sample management, contact us today.
What is Stability Storage?
Stability storage is the placement of samples into environmentally controlled chambers to determine how the quality of the substance or product varies with time under the influence of environmental factors such as temperature, humidity and light.
Stability chambers are used for research and product development work by pharmaceutical, medical device, life sciences, food manufacturing companies and others.
At Q1 Scientific, we offer state of the art environmentally controlled and monitored stability storage facilities to meet all ICH requirements and non-ICH requirements.
Why Stability Studies are required?
Stability studies are a critical component in drug and regulated product development.
Stability studies determine whether any physical, chemical or microbiological changes affect the efficiency and integrity of the final product, thereby ensuring that a pharmaceutical or regulated product is safe and effective, irrespective of where in the world it will be supplied.
A good stability study programme is required for the registration and commercialisation of any pharmaceutical or regulated consumer product. It is also essential in determining the shelf life of any product and an integral aspect of the product development process. It gives both regulators and consumers’ confidence that the product will perform ‘as expected’ from the date of manufacture through to the end of the product’s shelf life.
See our post outlining the main objectives of stability testing.
How do you test stability?
To test the stability of a drug or regulated product, samples need to be stored in environmentally controlled chambers as part of a stability study programme.
Samples are then pulled at various time points and tested to see whether there are any physical, chemical or microbiological changes that are likely to impact drug or product quality, safety and efficacy.
This diagram outlines the stability storage process our team goes through with each client:
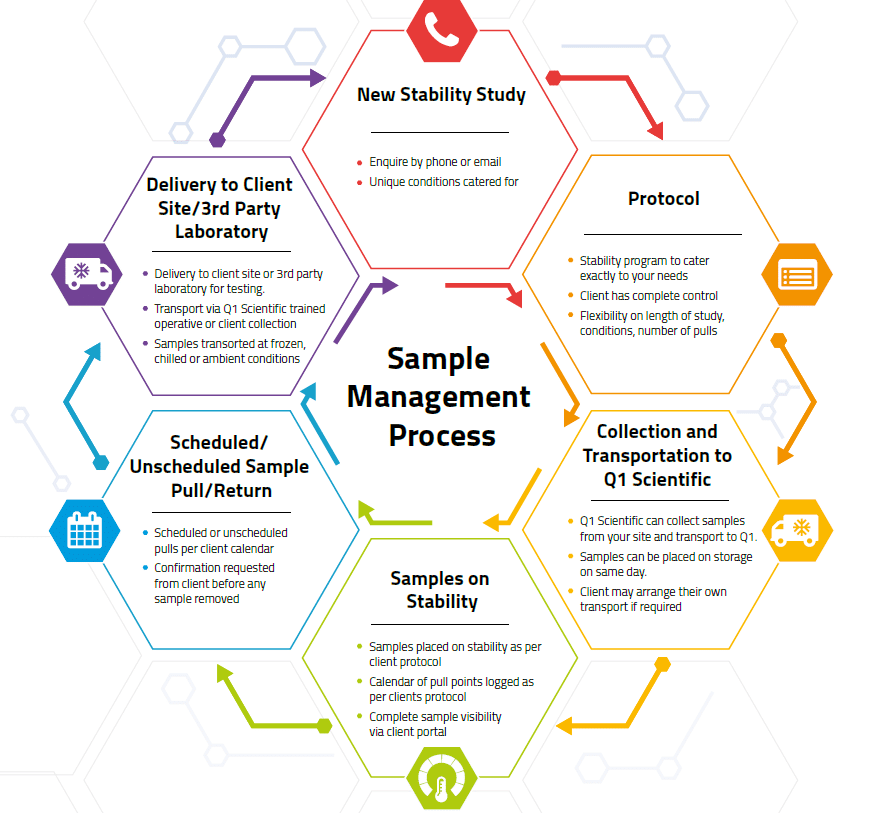
What are the stability zones and stability conditions?
To test stability, the International Council for Harmonisation (ICH) divides the world into five climatic zones based on a combination of temperature and relative humidity (RH). This division ensures that the differences in climatic conditions in the varying regions of the world are considered for stability studies. The five climatic zones are replicated in long-term stability studies to simulate the conditions worldwide that a drug substance or regulated product is subjected to.
Throughout the world, there are 5 different ICH Stability Zones:
| Climatic Zone | Type of Climate | Long term Stability Testing Recommended Conditions |
| Zone I | Temperate | 21°C/45%RH |
| Zone II | Mediterranean/Subtropical | 25°C/60%RH |
| Zone III | Hot, Dry | 30°C/35%RH |
| Zone IVa | Hot Humid/ Tropical | 30°C/65%RH |
| Zone IVb | Hot/ Higher Humidity | 30°C/75%RH |
Q1 Scientific has capacity to cater for all ICH climatic zones.
In addition to ICH conditions, Q1 Scientific also offer custom conditions to meet the specific storage requirements of any R&D project with options from -80°C storage up to +50°C with a full range of humidity control.

How long is stability testing?
- Long-term testing is generally carried out at 25°C/60%RH, for a minimum of 12 months
- Intermediate conditions, if required are generally 30°C/65%RH
- Accelerated testing is carried out at 40°C/75%RH, for a minimum of 6 months
See ICH Guidelines for recommendations around the length of stability testing.
What are the different types of stability testing?
The shelf life of a drug or regulated product is commonly estimated using long term and accelerated stability testing:
- Long term testing: Long-term stability testing is normally performed for a longer duration of the test period to allow significant product degradation under recommended storage conditions.
- Accelerated testing: Accelerated stability tests provide a means of comparing alternative formula-dons, packaging materials, and/or manufacturing processes in short-term experiments.
In addition, companies often perform:
- Reference / Retain sample testing: This is a usual practice for every marketed product for which stability data is required. Samples are retained to fulfil two purposes; firstly, to provide a sample for analytical testing and secondly to provide a specimen of the fully finished product. They serve as a record of the batch of finished product or starting material and can be assessed in the event of, for example, a dosage form quality complaint, a query relating to compliance with the marketing authorisation, a labelling/packaging query or a pharmacovigilance report.
- Thermal Cycling testing: Cyclic temperature stress tests are placed on the product so as to mimic likely conditions in marketplace storage. Thermal Cycling gives an indication as to how a product will react to adverse conditions, usually encountered during transportation. See our post on Thermal Cycling studies for more details.
How do you test accelerated stability?
The ICH defines this as:
Studies designed to increase the rate of chemical degradation or physical change of a drug substance or drug product by using exaggerated storage conditions as part of the formal stability studies
Most commonly, it is storage at 40°C +/- 2 degrees and Relative Humidity (RH) of 75% +/- 5% when the long-term stability storage condition for the product is 25°C +/- 2 degrees and RH of 60% +/- 5%.
This and other storage conditions are outlined in the ICH Guidelines for Stability Testing.
Accelerated stability tests provide a means of comparing alternative formula-dons, packaging materials, and/or manufacturing processes in short-term experiments. As soon as the final formulation and manufacturing process have been established, the manufacturer carries out a series of accelerated stability tests which will enable the stability of the drug product or medical device to be predicted. From this, its shelf-life and storage conditions can be determined. Real-time studies must be started at the same time for confirmation purposes. Suitable measures should be taken to establish the utilisation period for preparations in multi-dose containers, especially for topical use.
What is long term stability testing?
The ICH defines this as:
stability studies under the recommended storage condition for the re-test period or shelf life proposed (or approved) for labelling
Long-term stability testing is also referred to as room temperature stability storage condition testing. The most common storage condition is 25°C +/- 2 degrees and RH of 60% +/- 5%.
This and other storage conditions are outlined in the ICH Guidelines for Stability Testing.
Why is a forced degradation study carried out?
Forced degradation is an important part of the drug development process as it provides knowledge about the degradation chemistry of drug substances and drug products. This knowledge is used primarily to develop stability-indicating analytical methods but is also useful for other purposes such as formulation development, packaging development and the design of the official stability studies. As there is no formal regulatory guidance for forced degradation, it is recommended to use appropriate conditions to achieve 5-20% degradation.
Knowledge gained in forced degradation studies can facilitate improvements in the manufacturing process.
To find out more, see our post on Forced degradation studies for Drug Substances and Drug Products
What is photostability?
All companies developing or manufacturing pharmaceutical drugs, require a robust photostability testing process to ensure product quality and regulatory compliance. Inadequate testing can result in costly delays and lost revenue.
The intrinsic photostability characteristics of new drug substances and products should be evaluated to demonstrate that, as appropriate, light exposure does not result in unacceptable change.
The ICH Q1B guideline is the harmonised effort to standardise photostability testing on new pharmaceutical drug substances and drug products.
Testing is carried out on a single batch of material selected. Under some circumstances these studies should be repeated if certain variations and changes are made to the product (e.g. formulation, packaging). Whether studies should be repeated depends on the photostability characteristics determined at the time of initial filing and the type of variation and/or change made.
To learn more about creating the appropriate test conditions in accordance with ICH Q1B, please see our recent articles on Photostability testing theory and practice and Photostability Testing Guidelines of New Drug Substances.
What are reference and retain samples?
Reference samples are retained to fulfil two purposes; firstly, to provide a sample for analytical testing and secondly to provide a specimen of the fully finished product. They serve as a record of the batch of finished product or starting material and can be assessed in the event of, for example, a dosage form quality complaint, a query relating to compliance with the marketing authorisation, a labelling/packaging query or a pharmacovigilance report.
Samples may therefore fall into two categories:
- Reference sample: a sample of a batch of starting material, packaging material or finished product which is stored for the purpose of being analysed should the need arise during the shelf life of the batch concerned. Where stability permits, reference samples from critical intermediate stages (e.g. those requiring analytical testing and release) or intermediates, that are transported outside of the manufacturer’s control, should be kept.
- Retention sample: a sample of a fully packaged unit from a batch of finished product. It is stored for identification purposes. For example, presentation, packaging, labelling, patient information leaflet, batch number, expiry date should the need arise during the shelf life of the batch concerned.
For finished products, in many instances the reference and retention samples will be presented identically, i.e. as fully packaged units. In such circumstances, reference and retention samples may be regarded as interchangeable.
See our recent article on Reference and Retain Samples to find out more about the storage requirements.
What is Stability data?
The drug regulatory authority will require manufacturers to submit data on the stability of the product derived from tests on the final dosage. The tests must be completed on the product while in its final container and packaging. The data submitted is obtained from both accelerated and real-time studies. Published and/or recently obtained experimental supporting stability data may also be submitted, e.g. on the stability of active ingredients and related formulations.
Where the product is to be diluted or reconstituted before being administered to the patient (e.g. a powder for injection or a concentrate for oral suspension), “in use” stability data must be submitted to support the recommended storage time and conditions for those dosage forms.
With the approval of the drug regulatory authority, a tentative (provisional) shelf-life is often established, provided that the manufacturer has undertaken, by virtue of a signed statement, to continue and complete the required studies and to submit the results to the registration authority.
Post-registration the manufacturer must carry out on-going real-time stability studies to substantiate the expiry date and the storage conditions previously projected. The data needed to confirm a tentative shelf-life must be submitted to the registration body.
Once the product has been registered, additional studies are required whenever major modifications are made to the formulation, manufacturing process, packaging or method of preparation. The results of these studies must be communicated to the competent drug regulatory authorities.
Would you like to learn more about Stability Study design?
Following feedback from clients we have curated a new webinar series. This webinar series is designed for those working in the pharmaceutical, medical device and life sciences sectors with responsibility for designing and managing stability studies.
The webinars aim to provide an insight into stability study design and what it means for you. Pharmaceutical industry expert Dr. Mark Powell, shares his experience and address key questions in these free webinars.
Topics include:
- Forced degradation studies
- Managing drug-related degradation products
- Registration stability case study
- In-use stability
- Stability Studies throughout the product life cycle
- Designing a registration stability study from scratch
- Trending stability data
- Shelf life evaluation and extrapolation
- Temperature excursion and cycling studies
- Biopharmaceutical stability studies
- Stability study design – bracketing and matrixing
View previous Q1 Scientific stability study webinars
How can Q1 Scientific help you?
Q1 Scientific offer a complete stability sample management service and can organise and secure your sample inventory. We offer:
- ICH compliant and non-ICH stability storage chambers which are monitored 24/7, 365 days of the year and have built-in safeguards
- Custom conditions to meet the specific storage requirements of any project
- Fully validated systems and equipment
- A monitoring and alert system and emergency back-up generators
- A complete stability study sample management service with sample receipt and return documentation along with notifications for scheduled pulls for testing
- A secure cloud-based documentation solution for your sample management
- Temperature controlled transfer and transport
- Access to a team of highly experienced pharmaceutical industry experts who are trained in recording cGMP compliant data
With tightly controlled temperatures and humidity rates at our spacious cGMP facility, we can store and manage any samples over the full life cycle of your stability plans.
To discuss how we can help you contact the team today.